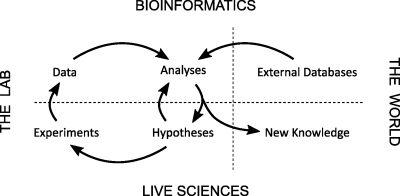
Die Methylierung von CpG Dinukleotiden in der DNA hat umfassende Folgen für die Transkription und bildet damit einen wichtigen Grundpfeiler für die Regulation der Genexpression. Einblicke in diese Methylierungsmuster setzen Programmierkenntnisse voraus und werden erschwert durch einen schwer zu durchschauenden Dschungel von Software. Das aktuelle Softwaretechnikprojekt macht es sich zur Aufgabe, diese Probleme in eine nutzerfreundliche Softwarelösung zu katalysieren, die schnelle und unkomplizierte Einblicke in die komplizierte Welt der DNA-Methylierung ermöglicht.
SWTP Teams BigTom und Biotique, Technische Hochschule Mittelhessen
"Modellierung und Analyse von Petrinetzen im Webbrowser mit Petrify"
Petrify ermöglicht die Modellierung und Analyse von Petrinetzen direkt im Webbrowser. Petrinetze sind ein mathematisches Modell zur Beschreibung von verteilten Systemen und parallelen Prozessen. Sie bestehen aus Stellen (Places), Transitionen (Transitions) und gerichteten Kanten (Edges), die das Verhalten dynamischer Systeme abbilden. Die Tokenverteilung in den Stellen repräsentiert den aktuellen Zustand des Systems.
"From Nose to Lungs: A Systems Biology Journey To Fight Infections"
The respiratory tract is the interface between the external environment and the human body, acting as the main entry point for numerous pathogens and commensal bacteria. The composition and homeostasis of these microbiomes profoundly impact the development of infectious diseases. Still, the contribution of many nasal microorganisms to human health remains undiscovered. The relationship between these microorganisms and the viral infection is also questionable. We are employing Systems Biology and Machine learning to decipher the connection between a diverse human microbiome and associated diseases caused by different stages of infection. While we lack the microbial information and uncultured microbes in vivo, genome-scale metabolic models (GEMs) can grant the key steps toward understanding the principles of microbes. Furthermore, GEMs can be valuable in simulating viral interactions and integrating these interactions within the host to identify vulnerabilities in the virus. These approaches open up a fascinating scenario of possible novel discoveries and hypotheses. Currently, there is no specific treatment for viral infections, whereas vaccination can effectively prevent pandemics. Antibiotic treatment is beneficial for bacterial infections, but the production of antibiotics is not increasing, and many microorganisms have developed antibiotic resistance. Medical control strategies for infectious diseases remain enormously important. The severity of global infections caused by hard-to-treat pathogens and viruses drives us to simulate infection scenarios in silico to explore alternative treatment options. This understanding may also open the way for the development and use of new drugs in the near future.
Dr. Reihaneh Mostolizadeh, Justus Liebig University Giessen and Max-Planck-Institute for Terrestrial Microbiology
Im ersten KoBIS dieses Jahres stellen die Studis des aktuellen Softwaretechnikprojektes ihre Python Library mit angebundenem Webservice - pyoSeq - vor. Ziel des Projektes war es, aufbauend auf einem früheren Projekt, bekannte Algorithmen der Bioinformatik zu implementieren, zu erklären und die Ergebnisse der Algorithmen zu visualisieren, um eine Ressource zu schaffen, mit deren Hilfe Studis die Algorithmen besser verstehen können. pyoSeq kann dabei sowohl von Studis genutzt, als auch von Dozierenden in der Lehre eingesetzt werden. pyoSeq bietet zusätzlich die Option, eigene Implementierungen der enthaltenen Algorithmen auf mögliche inhaltliche Fehler zu testen.
Titanis AG (Softwaretechnikprojekt Wise 24/25), THM Gießen
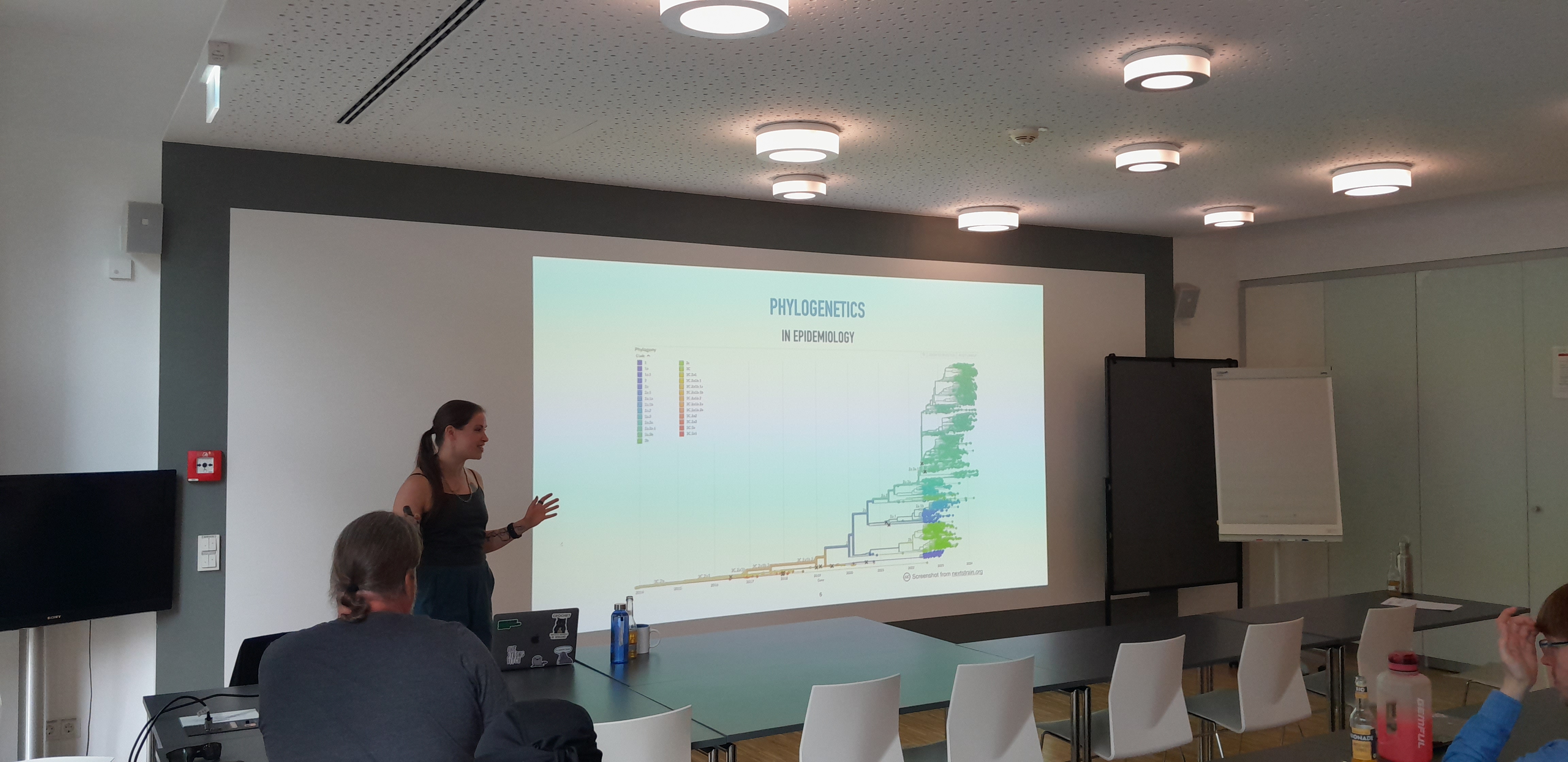
"Mind the gaps: the many pitfalls of phylogenetic inference"
Phylogenetic tree reconstruction and multiple sequence alignment (MSA) are fundamental tasks in phylogenetics that precede most other types of genetic sequence analysis. These two tasks should ideally be performed jointly since trees and alignments are inherently circularly dependent. In reality, however, the two structures are more often than not inferred sequentially as independent objects. Moreover, aligners infer MSAs that contain gaps meant to represent insertion and deletion events throughout evolutionary history. On the other hand, most standard likelihood-based phylogenetic tree reconstruction tools treat the MSA as data and filter out gappy columns because they cannot be included in the phylogenetic likelihood computation.
One way to solve this disparity is to include evolutionary models that explicitly account for insertions and deletions in the likelihood computation. Such models would allow us to use all available data in inference without hacking our way around handling gaps. Unfortunately, most evolutionary models that handle indels meaningfully are computationally intractable, which adds further hurdles to the already complex task of phylogenetic inference.
In this talk, we will discuss the status quo in phylogenetics, its many hidden pitfalls, and the current work in Maria Anisimova's group on an efficient joint inference method.
Dr. Julija Pecerska, Zurich University of Applied Sciences (ZHAW), Life Sciences, Bioinformatics
"Deep Learning basierte Generierung von antimikrobiellen Peptiden. Auf der Suche nach neuen Antibiotika."
Das Problem der antimikrobiellen Resistenz gewinnt zunehmend an Bedeutung. Schätzungen zufolge werden bis 2050 jährlich mehr als 10 Millionen Menschen an den Folgen der Antibiotikaresistenz sterben. Aktuelle Forschungen zeigen, dass der Einsatz von Deep Learning, insbesondere mit generativen Modellen, nicht nur dabei helfen kann neue Antibiotika zu finden, sondern auch die Entwicklungszeit zu verkürzen und gleichzeitig die Erfolgsraten zu erhöhen. In diesem Vortrag wird auf aktuelle Techniken zur Generierung von antimikrobiellen Peptiden eingegangen.
B. Sc. Lukas Beierle, Technische Hochschule Mittelhessen / Justus-Liebig Universität Gießen
"Fungi that Infect Humans"
Pilze müssen vier Kriterien erfüllen, um den Menschen zu infizieren: Wachstum bei menschlicher Körpertemperatur, Umgehung oder Durchdringung von Oberflächenbarrieren, Lyse und Absorption von Gewebe sowie Resistenz gegen die Immunabwehr, einschließlich erhöhter Körpertemperaturen. Die Morphogenese zwischen kleinen, runden, abtrennbaren Zellen und langen, zusammenhängenden Zellen ist der Mechanismus, mit dem Pilze das Problem der Fortbewegung um oder durch Wirtsbarrieren lösen. Die Sekretion von lytischen Enzymen und Aufnahmesysteme für die freigesetzten Nährstoffe sind notwendig, damit ein Pilz menschliches Gewebe ernähren kann. Schließlich hat sich das starke menschliche Immunsystem in der Interaktion mit potenziellen Pilzpathogenen entwickelt, so dass nur wenige Pilze alle vier Bedingungen für einen gesunden menschlichen Wirt erfüllen.
Paradoxerweise haben die Fortschritte der modernen Medizin Millionen von Menschen durch die Unterbrechung der Immunabwehr neu anfällig für Pilzinfektionen gemacht. In diesem Artikel wird untersucht, wie verschiedene Mitglieder von vier Pilzgruppen unterschiedliche Strategien anwenden, um die vier Kriterien für eine Infektion des Menschen zu erfüllen
"3D Finite-Elemente-Simulation von Diffusion und Zellzyklus in Tumorgewebe (Deutsch)"
Im Rahmen eines Softwaretechnik-Projektes wurde eine Software entwickelt, welche den Transport von Stoffen innerhalb eines Biologischen Systems und das Verhalten der sich darin befindenden Zellen simuliert. Das Thema wurde eingegrenzt auf Sauerstoff und Glukose in Brustkrebs-Gewebe.
"Probleme und Lösungen bei Bindeaffinitäts- und Kreuzreaktionsvorhersagen von MHC I T-Zell Epitopen (Deutsch)"
T-Zell Epitope sind in der immunolgischen Forschung von besonderem Intersse. Vor Allem bei Autoimmunerkankungen, Allergien, Epitop-basierten Vakzinen und Kreuzreaktionen.
Aktuell werden viele Anwendungen auf basis von maschinellem Lernen entwickelt um beispielsweise die Bindungsaffinität oder ein Potential für Kreuzreaktionen der Epitope vorherzusagen.
In diesem Vortrag werden die aktuellen Projekte der AG Cemic zu diesem Thema vorgestellt und auf die dabei enstandenen Probleme, sowie deren Lösungen eingegangen.
"Haplotypenbasierte Leistungsvorhersage in Gerste (auf Deutsch / in German language)"
Die Verfügbarkeit von Markerdaten hat die Möglichkeiten zur Selektion in der Pflanzenzüchtung erweitert: Sie können verwendet werden, um den genomischen Wert von Individuen zu schätzen und die Individuen mit dem besten genomischen Wert auszuwählen (Genomic Selection/Genome-wide Prediction). Neuere Verfahren betrachten nicht mehr die Effekte einzelner Marker, sondern teilen das Genom in verschiedene Abschnitte, sogenannte Haplotypblöcke, für die dann Effekte geschätzt werden. Die Erfolgsaussichten dieser Vorgehensweise hängen dabei vom untersuchten Merkmal, aber auch von der Methode der Erstellung der Haplotypblöcke ab. Außer zur genomischen Selektion können Haplotypblöcke aber auch zur Visualisierung von Regionen auf dem Chromosom genutzt werden, die einen positiven oder negativen Effekt auf das Zielmerkmal haben.
Dr. Carola Zenke-Philippi, Institute for Agronomy and Plant Breeding II, Justus Liebig University
"Meeting link"
https://thm-de.zoom.us/j/3269142051
Meeting ID: 326 914 2051
One tap mobile
+496971049922,,3269142051# Germany
Dial by your location
+49 69 7104 9922 Germany
Meeting ID: 326 914 2051
Find your local number: https://thm-de.zoom.us/u/cc3X4Mhpcn
You can either subscribe to the THM-mailinglist "kobis-mni" where the login credentials will be send one day in advance:
https://lists.thm.de/mailman/listinfo/kobis-mni
...or check this site on the presentation day.
,
"Wie erhalte ich Zugang zur Präsentation?"
Entweder durch Einschreibung in die THM-Mailingliste "kobis-mni", über welche die Zugangsdaten verschickt werden:
https://lists.thm.de/mailman/listinfo/kobis-mni
...oder hier, auf dieser Seite am Präsentationstag.
Land plants make up most of today's biomass, they produce oxygen, fix carbon and it was their evolution that made life on land as we know it possible. The history of their development takes us back to the emergence of eukaryotes, followed by the photosynthesising plastid and the subsequent changes in the atmosphere and constantly rising complexity.
Coming from salt water, algae began to adapt to life in fresh water and along the lineage of the Streptophyta further to drought. 500 million years ago, the earth was bare rock and provided a largely hostile environment for life. Adapted to drought and irradiation, however, bryophytes managed to colonise the land, paving the way for lycophytes, ferns and eventually seed plants to green up the earth by their rapid diversification
Mona Schreiber, Philipps-University Marburg
"Meeting link"
Topic: KoBIS 07.12.2021: The Greening Ashore
Time: Dec 7, 2021 04:00 PM Amsterdam, Berlin, Rome, Stockholm, Vienna
Meeting ID: 326 914 2051
One tap mobile
+496971049922,,3269142051# Germany
Dial by your location
+49 69 7104 9922 Germany
+1 206 337 9723 US (Seattle)
Meeting ID: 326 914 2051
Find your local number: https://thm-de.zoom.us/u/kvoxJCRJy
Der heutige Vortrag wird auf das Frühjahr 2022 verschoben.
Todays talk is postponed to 2022.
,
"Haplotypenbasierte Leistungsvorhersage in Gerste (auf Deutsch / in German language)"
Die Verfügbarkeit von Markerdaten hat die Möglichkeiten zur Selektion in der Pflanzenzüchtung erweitert: Sie können verwendet werden, um den genomischen Wert von Individuen zu schätzen und die Individuen mit dem besten genomischen Wert auszuwählen (Genomic Selection/Genome-wide Prediction). Neuere Verfahren betrachten nicht mehr die Effekte einzelner Marker, sondern teilen das Genom in verschiedene Abschnitte, sogenannte Haplotypblöcke, für die dann Effekte geschätzt werden. Die Erfolgsaussichten dieser Vorgehensweise hängen dabei vom untersuchten Merkmal, aber auch von der Methode der Erstellung der Haplotypblöcke ab. Außer zur genomischen Selektion können Haplotypblöcke aber auch zur Visualisierung von Regionen auf dem Chromosom genutzt werden, die einen positiven oder negativen Effekt auf das Zielmerkmal haben.
Dr. Carola Zenke-Philippi, Institute for Agronomy and Plant Breeding II, Justus Liebig University
"How to get access to the presentation?"
You can either subscribe to the THM-mailinglist "kobis-mni" where the login credentials will be send one day in advance:
https://lists.thm.de/mailman/listinfo/kobis-mni
...or check this site on the presentation day.
,
"Wie erhalte ich Zugang zur Präsentation?"
Entweder durch Einschreibung in die THM-Mailingliste "kobis-mni", über welche die Zugangsdaten verschickt werden:
https://lists.thm.de/mailman/listinfo/kobis-mni
...oder hier, auf dieser Seite am Präsentationstag.
"Introducing the Component Library Engineer as future job description in systems biology (English)"
When comparing mathematical modeling projects in systems biology to those in industrial applications, one major difference in the general workflow becomes apparent: Virtually all large-scale industrial models are based on well-tested and carefully designed libraries of standardized components, which can then be used for a wide variety of applications. Although there exists no unifying theory for biological systems that is comparable to laws for mechanical or electrical systems, my research has shown that the creation of robust, modular component libraries is a viable and much-needed approach to building multi-scale, cardiovascular models. But who should create these libraries? The requirements for this task can neither be found in traditional software engineering nor systems biology courses alone. Hence, I propose a new educational specialization and job description: the Component Library Engineer.
"Softwaretechnikprojekt: Modelica-Programmierung überall mit dem Modelica-Pipe-Editor (Mo|E) und dem Language Server Protocol (English)"
Das Ziel des Projektes war die einfache und komfortable Programmierung mathematischer Modelle mit Modelica ohne schwergewichtige IDE zu ermöglichen. Der Modelica-Pipe-Editor Mo|E, der einen Serverprozess für die Kommunikation mit dem OpenModelica Compiler aus einem Texteditor heraus bereitstellt, sollte dazu neu entwickelt und/oder verbessert werden. Dafür sollte Microsofts Language Server Protocol verwendet werden, um die Kommunikation mit Client-Plugins z.B. in VS Code oder Atom zu vereinfachen. Der Entwicklungsprozess sollte agil gestaltet werden mit viel Freiheit in der Wahl von verwendeten Tools und Plattformen.
Ilmar Bosnak, Conrad Lange, Manuel Wächtershäuser, THM Gießen
"Software engineering project: Modelica development everywhere with the Modelica-Pipe-Editor (Mo|E) and the Language Server Protocol"
The goal of this student project was to allow easy and comfortable programming of mathematical models in Modelica without heavyweight IDEs. It is based on the existing Modelica-Pipe-Editor (Mo|E), which provides a server process for the communication with the OpenModelica compiler from a text editor. The students refactored Mo|E using the Language Server Protocol to facilitate the communication with plugins for clients such as VS Code or Atom. The development process used agile methods and the students had the freedom to choose their favorite tools and platforms.
Ilmar Bosnak, Conrad Lange, Manuel Wächtershäuser, THM Gießen
"Aufbau einer Datenbank für kreuzreaktive T-Zell Epitope (Deutsch)"
Kreuzreaktive T-Zell Epitope sind von besonderem Interesse, gerade bei der Erforschung von Allergien, Epitop-basierten Impfungen oder sogenannten Kreuzreaktionen.
Zur Zeit lassen sich kreuzreaktive T-Zell Epitope nur sehr schwer und kostenintensiv im Labor validieren, bzw. untersuchen. Daher ist es sinnvoll in silico Vorhersagetools mittels maschinellem Lernens zu entwickeln.
Aktuell werden bestehende Vorhersagen zur Kreuzreaktivität von T-Zell Epitopen nur auf Basis von Sequenzhomologie durchgeführt. Das Ziel dieses Projektes ist es, zusätzlich sequenzbasierte Marker für di Kreuzreaktivität für T-Zell Epitope herauszuarbeiten, diese Daten in Form einer Datenbank bereitzustellen und damit bestehende Pipelines zur Bestimmung von kreuzreaktiven T-Zell Epitopen zu verbessern.
Im Rahmen dieses Vortrages wird die Arbeit, bzw. der Fortschritt zum Aufbau einer Datenbank für diese Epitope vorgestellt.
Feature selection is an important step for many applications of data in machine learning. The 'right' choice of features can improve classification while reducing training time. Many different selection methods are available. For a more suitable way in selecting features, multiple selection methods can be combined in an ensemble. Features are then selected by majority vote.
This talk will present a feature selection ensemble written in Python with the usage of different approaches, including: filters, embedded, wrappers and various others.
"Softwaretechnikprojekt: Bildbasierte Qualitätserkennung in der Steinplattenproduktion mittels neuronaler Netze"
Während eines Softwaretechnikprojekts haben sich zwei Teams damit beschäftigt, ein künstliches neuronales Netz zur Qualitätserkennung von Terassenplatten zu entwickeln. Dazu wurden zuerst Skripte zur Bildaufnahme entwickelt, die dadurch entstandenen Bilder bearbeitet, künstlic hvermehrt und anschließend in mehreren Durchläufen verschiedene Netze trainiert und evaluiet. Für das Frontend wurde ein GUI geschaffen.
Vortragssprache ist Deutsch, es können anschließen dFragen auf Englisch gestellt werden.
Presentation is hold in German, questions can be asked in English, too.
M. Albaghshawngy, B. Blattmann, S. Bock, P. Georgiew, R. Hohl, C. Ifland, B. Riegel, M. Rinker, P. Schlag, M. Weller, Technische Hochschule Mittelhessen
"Co-occurrence prediction of Transcription Factor Binding Sites based on ATAC-seq footprinting data"
Transcription factor (TF) binding can be referred from ATAC-seq data by an approach defined as digital genomic footprinting. We recently introduced a software named TOBIAS (Transcription factor Occupancy prediction By Investigation of ATAC-seq Signal), able to perform such analysis and to quantify TF-binding activity on multiple samples and conditions in a global scale.
However, TOBIAS does not provide a function to unravel the co-occurrence of TFs at genomic loci such as enhancers or promotors. This is of high importance when investigating the complex mechanisms of transcriptional regulation.
In this KoBis talk, I will introduce a method to predict TF co-occurrences and the comparison between different conditions by an adaptation of a market basket analysis.
Vanessa Heger, Max-Planck-Institut Bad Nauheim, Justus-Liebig Universität Gießen, Technische Hochschule Mittelhessen
All data has hidden facets. Whether it is the space of representation or the different data, different constraints determine how we think about it. In this talk, these constraints from different fields of knowledge will be addressed, in particular bioinformatics approaches and workflows. The focus of data representation will shift from sequence to structure, usability and diversity, and finally to rules for better quality.
"Beyond accessibility: ATAC-seq footprinting unravels kinetics of transcription factor binding during zygote genome activation"
While digital genomic footprinting analysis of ATAC-seq data can theoretically enable investigation of transcription factor (TF) binding, the lack of a computational method implementing both footprinting and downstream analysis has hindered the widespread application of this method. By this talk, I present TOBIAS, a comprehensive footprinting framework enabling genome-wide investigation of TF binding dynamics for hundreds of TF simultaneously. As a proof-of-concept, TOBIAS is used to unveil complex TF dynamics during zygotic genome activation (ZGA) in both humans and mice. In particular, TOBIAS is used to illustrate the activity of the TF Dux, its potential to bind repeat elements and its potential to induce expression of novel genetic elements.
Prof. Dr. Mario Looso, Max-Planck-Institut für Herz- und Lungenforschung, Bad Nauheim
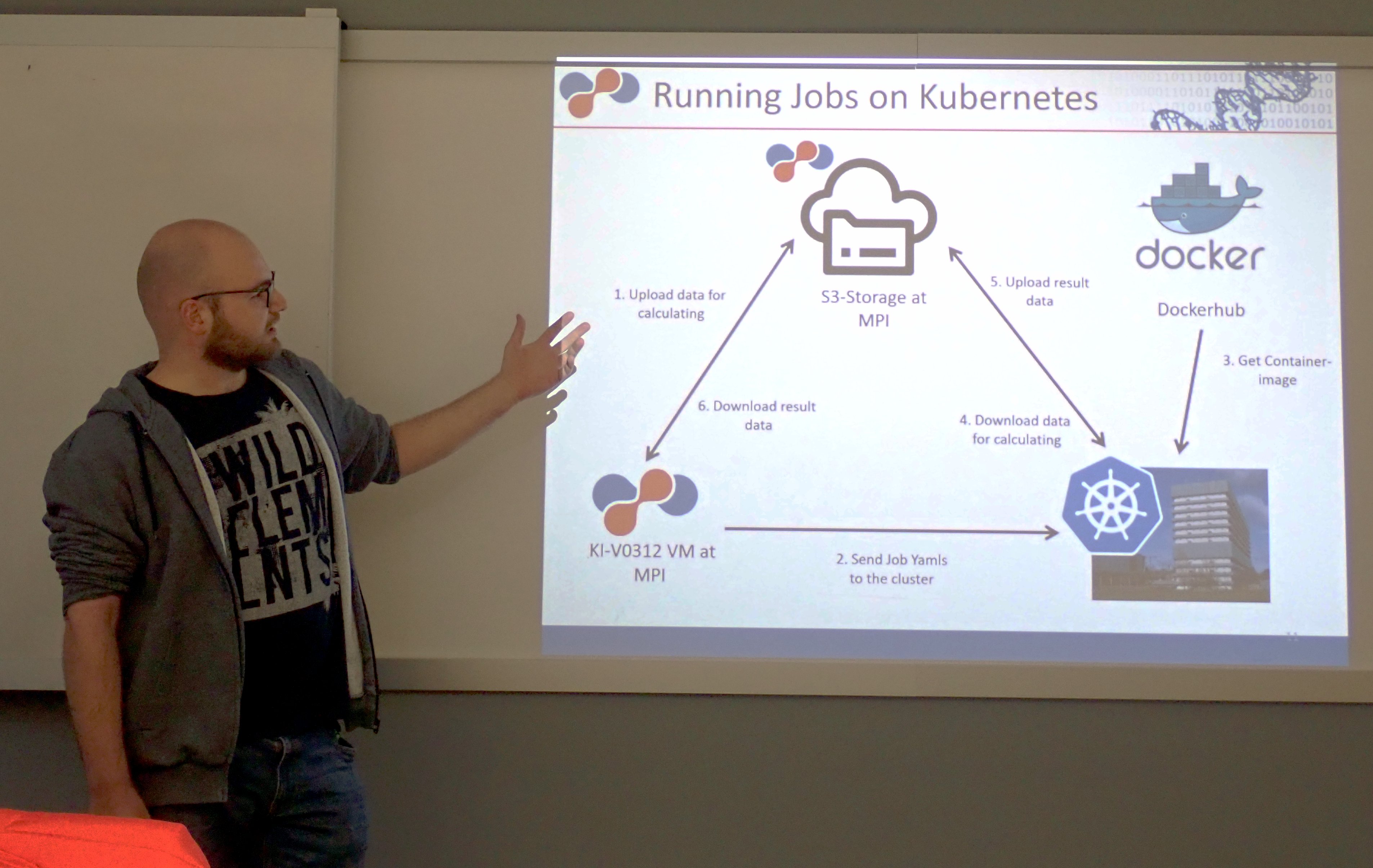
"deNBi Cloud: Mapoks and macsek, frameworks to manage nextflow pipelines on kubernetes"
Costs of data acquisition in the field of applied bioinformatics are decreasing. Therefor the costs for data analysis can become a bottleneck due to a lack of computing resources. Cloud computing is a solution for cost-effective computing resources on demand. The german network for bioinformatics (deNBi) provides cloud computing resources on systems such as Kubernetes. In order to facilitate the transmission of bioinformatics pipelines to the cloud, two methods have been developed that enable the deployment of pipelines on the deNBi Gießen Kubernetes cluster. The first method MAPOKS (Managing Pipelines on Kubernenets with an S3-Storage) was developed for authorized users of a Kubernetes cluster.
With the second method MACSEK (MAnaging Computing SErvices on Kuberntes) a framework was developed that enables an open computing service on a Kubernetes-cluster. These frameworks essentially simplify the allocation of cloud resources for individual bioinformatics software and pipelines and will path the road for a widespread utilization of such resources in the future.
Philipp Goymann, Max-Planck-Institut für Herz- und Lungenforschung, Bad Nauheim
Sequencing-based analysis of microbiota is a crucial tool for many biological and medical disciplines to investigate microbial diversity that would otherwise be missed by culture-based approaches. Shifts in microbial composition can lead to phenotypic changes of the host, like obesity in mice, Clostridium difficile infection in human or the development of Parkinson's disease. Shifts can be induced via dietary changes or addition of (anti-), pre- or probiotics, which might open up routes for beneficial host modulations in e.g. human health.
Unfortunately, microbiome data is typically very noisy - not only due to technical biases ranging from sample handling to sequencing platform of choice, but also due to many biological confounding factors like diet, age, ethnicity, hygiene or country.
My talk will cover statistical concepts of state of the art tools to disentangle different effects and to quantify microbial ecology sequencing data applied to an ongoing microbiome project investigating the role of the gut microbiome in childhood leukemia development.
Prof. Dr. Stefan Janssen, Justus-Liebig Universität Gießen
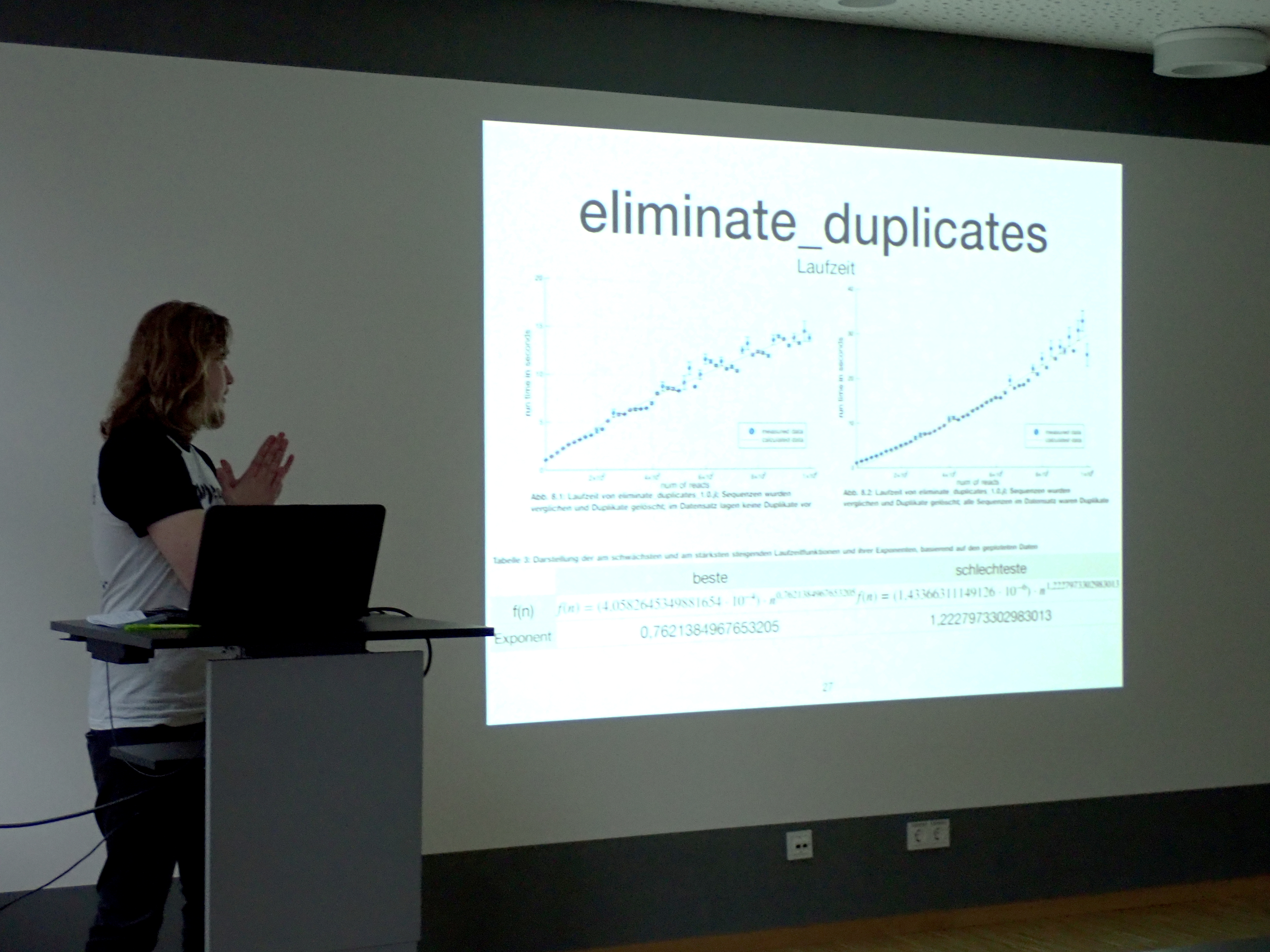
"eliminate_duplicates – Ausschalten von Read-Duplikaten innerhalb von Alignments von single-end-Reads"
Reads sind Basen-Sequenzen, die bei der Sequenzierung abgelesen werden und je ein Sequenz-Fragment der Ausgangsprobe darstellen. Read-Duplikate können auf vielfache Art entstehen, bevor die Sequenzen als Reads abgespeichert werden: Ausgangssequenzen können mehrfach in der Probe vorliegen, bei der PCR können Sequenzduplikate entstehen und während der Illumina-Sequenzierung kann ein Cluster als mehrere identifiziert werden, wodurch optische Duplikate entstehen.
Aligniert man die Reads an ein entsprechendes Referenzgenom, so lassen sich diese Duplikate identifizieren und für weitere Analyseschritte eliminieren. Dies kann dazu dienen, dass eben diese folgenden Analysen weniger zeit- und speicheraufwändig sind. Herkömmliche Tools sind zumeist auf die Duplikatelimination von paired-end-Reads ausgelegt und betrachten unzureichende Kriterien, werden sie auf single-end-Reads angewendet, was zu falsch-positiven Eliminationen führt.
Um diese zu reduzieren wurde ein Algorithmus entwickelt, welcher speziell auf Elimination von Read-Duplikaten in single-end-Daten ausgelegt ist.
Maximilian Nocke, Technische Hochschule Mittelhessen
"ASA³P: automatic and scalable assembly, annotation and higher level analysis of closely related bacterial isolates"
Bakterien sind winzige Mikroorganismen mit vielfältigen und faszinierenden Eigenschaften, welche im Laufe der Evolution jeden bekannten Ort dieses Planeten erfolgreich besiedelt haben; insbesondere auch den menschlichen Körper. Viele fungieren als nützliche Symbionten und sind für unser Wohlbefinden sehr wichtig. Einige Wenige verursachen jedoch teils schwere Krankheiten. Viele bakterielle Krankheitserreger sind immer häufiger mit zahlreichen Antibiotika-Resistenzen ausgestattet. Laut einer Studie der WHO könnten bis zum Jahr 2050 alle bekannten Antibiotika wirkungslos werden und millionen Menschen weltweit erneut an längst besiegten Krankheiten sterben. Andererseits lassen sich Bakterien dank der modernen Biotechnologie in vielfältiger Weise nutzen. Die Bandbreite spannender Anwendungsgebiete reicht hierbei von der Produktion von Medikamenten, Spezialchemikalien und Biokraftstoffen über den Abbau von Abfällen und Toxinen bis hin zur Korrosionsbekämpfung.
Dank der weltweiten Etablierung von „Next-“ und „Third-Generation-Sequencing“-Methoden ist die Sequenzierung bakterieller Genome zur Routine geworden und wandelt sich derzeit unaufhaltsam zu einer Big-Data-Wissenschaft. Um mit diesen Entwicklungen Schritt zu halten und aus den riesigen Datenmengen neue Erkenntnisse zu ziehen, müssen moderne Algorithmen und Datenbanken der Bioinformatik in intelligenten Workflows passgenau mit einander verbunden werden. Damit diese von Wissenschaftlern und Medizinern weltweit genutzt werden, müssen sie gleichermaßen skalierbar, portierbar und nutzerfreundlich zu bedienen sein.
Zur gezielten und umfassenden Analyse bakterieller WGS-Daten wurde die Analyse-Software ASA³P entwickelt. ASA³P unterstützt die Verarbeitung von Rohsequenzierdaten der derzeit wichtigsten Sequenzier-Systeme. Neben umfassender Qualitätskontrollen werden assemblierte Sequenzen an Referenzgenomen ausgerichtet und anschließend annotiert. Darauf basierend werden sämtliche Genome ausführlich charakterisiert, was derzeit die taxonomische Klassifizierung, MLST-Typisierung, Detektion von Antibiotikaresistenz- und Virulenzgenen als auch die Detektion von SNPs umfasst. Abschließende komparative Analysen wie die Berechnung des Core- und Pan-Genoms sowie die Erstellung phylogenetischer Bäume erlauben einen ersten Einblick in die Verwandtschaftsgrade der Organismen. Sämtliche daraus resultierende Ergebnisse werden dem Benutzer in Standard-Dateiformaten der Bioinformatk angeboten und zusätzlich in modernen HTML5-basierten Dokumenten mit interaktiven Visualisierungen aufbereitet. ASA³P wird als lokal ausführbare Docker-Variante und zusätzlich als hoch-skalierbare OpenStack-basierte Cloud-Computing-Variante angeboten. Mithilfe der Pipeline ist die portable, skalierbare Prozessierung, Analyse und umfassende Charakterisierung hunderter bakterieller Genome innerhalb weniger Stunden realisierbar.
Oliver Schwengers, Justus-Liebig Universität Gießen
"Sequenzierfehlererkennung in NGS-Daten mittels Wavelet-Basis und Random Forest Klassifikator"
Insbesondere die Erkennung von Punktmutationen mit geringer Abundanz in NGS-Daten stellt eine Herausforderung dar, weil Punktmutationen schwer von Sequenzierfehlern unterscheidbar sind. Diese Sequenzfehler sollten erkannt und aus der Datenmenge entfernt werden, um sicherere Analysen zu ermöglichen. MultiRes ist ein Algorithmus der k-mere unterschiedlicher Größe verwendet. Er basiert auf einer Wavelet-Basis und nutzt das Klassifikationsverfahren Random Forest, um fehlerhafte k-mere von Punktmutationen zu unterscheiden. Im Rahmen dieses Vortrages wird dieses Verfahren vorgestellt.
"kASA: Taxonomic Identification of Metagenomic Data on a Notebook"
The identification of metagenomic sequences has gained importance in many areas of life sciences. Therefore, a large number of tools have been developed in recent years. However, most of these tools either consume a large amount of RAM, which is only available in high performance computer clusters, or provide insufficient quality of classification results.
Here we have developed kASA, a k-mer based tool capable of assigning and profiling metagenomic sequences with high computational power and user definable memory requirements.
By using new technologies as well as custom-made algorithms and data structures we achieve a very high sensitivity and precision without compromises on a notebook.
Breast and ovarian cancers pose huge and still largely unsolved challenges for the medical profession. Most women with screening-detected breast cancer do not receive a clear treatment benefit, indicating that these cancers are still not being detected early enough. The less common ovarian cancer is often not diagnosed until in an advanced stage, resulting in a five-year survival rate of less than 40%. New tests based on genomic biomarkers have the potential to offer, for the first time, much earlier detection and diagnosis of ovarian cancers as well as improvements to routine breast cancer screening.
The EU FP7 EpiFemCare consortium set out to improve patient outcomes for breast and ovarian cancer by developing and validating a series of blood tests based upon DNA methylation technology. These “liquid biopsies” will facilitate both early detection and prediction of therapeutic outcome of breast and ovarian cancer. We report here how the implementation of a specific bioinformatics analysis of Reduced Representation Bisulfite Sequencing (RBBS) data to detect cancer-specific methylation regions helped to develop an assay applicable in clinical diagnostics.
"Chemical diversity in Xenorhabdus and Photorhabdus: interpretable machine learning for natural product research"
Xenorhabdus and Photorhabdus are soil dwelling bacteria which produce a number of shared natural products (NPs) and occupy very similar ecological niches. They are found worldwide in association with nematodes of the genera Heterorhabditis and Steinernema, respectively, and, despite their specificity to the nematode hosts, bacteria-nematode pairs may be isolated from the same geographical location. NP research is continually encountering the problem of the best way to prioritise new metabolites: one common way to do this is using genomic information to identify gene clusters that often produce bioactive compounds, and subsequently activating “silent” clusters to specifically stimulate production of certain metabolites; however, in absence of genetic information, this becomes increasingly difficult.
Here, we turn to the field of machine learning, and construct a model to classify bacterial samples using a gradient boosting algorithm. While high-performing models tend to come in the form of black boxes, recently-developed feature attribution approaches open up the possibility of building state-of-the-art models, while at the same time minimising the trade-off between performance and interpretability. The use of these tools allows us to identify a compound that may be used as an extremely good predictor of the genus of a given sample. At the same time, we observe that geographical metadata does not seem to contribute to the classification. These results point towards a dependence of the bacteria upon the nematode in the environment, an area that has not been widely investigated due to the relative simplicity of growing them in the lab.
César A. Parra-Rojas, Ph.D., Systems Medicine of Infectious Diseases Frankfurt Institute for Advanced Studies, Frankfurt am Main, Germany
"Using machine learning methods for (bio-)diversity estimation of intricate data"
There are prosperous possibilities in biodiversity research for machine learn- ing methods. Especially for sophisticated methods like deep neural networks, which are currently trending, for example, in image recognition and natural speech processing. We apply deep neural networks on the task of diversity esti- mation on intricate data, where using elementary indices of (bio-)diversity can be troublesome. With a multi omics view in mind, we explore feasible network architectures for datasets of gene expression and metabolomics data. All eval- uated architectures culminate in an hourglass shape with a bottleneck. Where the size of the bottleneck represents the inherent complexity and diversity of the probed data. A way to relate these estimates thus obtained to other diversity indices is still pending
Christian Thomae Viegas, Fachbereich MNI, Technische Hochschule Mittelhessen
"Prediction of antiviral peptides with various machine learning algorithms"
Most viral infections are still extremly difficult to treat. Many existing drugs only relief the symptoms, but offer no cure. Antiviral peptides (AVP) could be used prophylactically before infection or afterwards. They are very common in nature, working as a first line of defence in many organisms. Synthesis and assessment of antiviral action on the other hand are very costly and time-consuming in a laboratory environment. Well trained machine learning algorithms can rapidly predict AVPs from sequence information. In combination with exponentially growing, dedicated sequence-databases an in silico identification of potential AVPs can be established. Promising candidates can then be subjected to further testing in a wet-lab environment. At first, indicative features characterising AVPs have to be selected, based on classification properties. Those can be from the chemical, physical or biological domain. Different methods will be applied to reduce the high dimensional featurespace to a smaller feature-set covering most of the relevant information about AVPs. This is a crucial step, as these features are used for training and define the classifiers. A pipeline of different machine learning approaches will be set up next, combining support vector machines, artificial neural networks and random forest. Furthermore, hyperparameters steering the classification algortithms will also be subjected to optimasation. A unified score weighting the outcome of different algorithms is used as an overall prediction-score. For that matter all subtleties of the different approaches will have to be considered. Finally, extensive laboratory validation will be conducted with a random representative set of predicted AVPs and peptides, predicted to have no (antiviral) effect.
Die Teams des Softwaretechnik-Praktikums stellen die Ergebnisse ihres Projekts vor. Aufgabe war es, eine Parrot Bebop 2 Drohne mittels einer KI autonom fliegen zu lassen. Die Teams entwickelten eine Fernsteuerung mit Gamecontroller und eine KI die selbstständig einer Bodenmarkierung folgt. Zusätzlich erkennt die Drohne verschiedene Ereigniskarten und führt dazu gehörende Flugmanöver durch.
Studenten des Softwaretechnik-Praktikums, Fachbereich MNI, Technische Hochschule Mittelhessen
"CrossCorrelationPredictor – Pipeline für Kreuzreaktivitätsvorhersage von Proteinen"
Im Rahmen eines Praktikums wurde sich mit Kreuzreaktivitätsvorhersage beschäftigt. Wie kürzlich herausgefunden wurde, gibt es virale Infektionen, die gegen Allergien oder andere nicht-verwandte Infektionen schützen können. Grund dafür sind Kreuzreaktivitäten zwischen Proteinen. Das heißt, zwei unterschiedliche Antigene reagieren auf den gleichen Antikörper in gleichem Maße. Somit kann das Wissen über bestehende Kreuzreaktivitäten helfen, Krankheiten besser zu verstehen und zu behandeln. Zum Zweck der Kreuzreaktivitätsvorhersage wurde eine Pipeline entwickelt, die Epitop-Vorhersagen von einer Protein-Datei und einer Protein-Datenbank macht und diese mittels Alignment und Cross Entropy miteinander vergleicht.
"GraphTeams: a method for discovering spatial gene clusters in Hi-C sequencing data"
Gene clusters are sets of genes that have associated biological function. In many organisms, instances exist where such genes are also locally close to each other in the genome, i.e., their positions fall within a narrow region on the same chromosome, despite the fact that genomes regularly undergo mutations such as genome rearrangements, gene- or segmental duplications, and gene insertions/deletions that disrupt gene neighborhoods. Because the function of many genes has yet to be understood, a popular approach for identifying gene clusters is to work its way backwards, starting with the investigation of conserved gene proximity in genomes of a reasonably phylogenetically diverse set of species.
In recent years, a new next-generation sequencing protocol called high-throughput chromosome conformation capture (Hi-C) has become widely popular that offers novel, cost-effective means to study the spatial conformation of chromatin in cells. The spatial arrangement of chromatin plays a crucial role in gene expression and exhibits conserved conformation across related species. In the presented work, data gathered from Hi-C sequencing experiments is used to study spatial gene clusters. These are sets of genes with associated functionality that exhibit close proximity to each other in the spatial conformation of chromosomes across related species.
In the presented work, a new gene cluster model is developed that is capable of handling spatial data. The model generalizes a popular computational model for gene cluster prediction from sequences to graphs. Following previous lines of research, the model, called δ-teams with families, is capable of handling of gene duplicates, which is particularly suitable for comparing spatial data of divergent species.
With GraphTeams, a fully automated workflow for discovering gene clusters in Hi-C data has be developed and applied to biological data to infer intra- and interchromosomal gene cluster candidates.
Technical developments in next-generation sequencing as well as decreasing prices have enabled various sequencing projects including the large genomes of higher eukaryotes. The two most popular and efficient methods for whole-genome sequencing are the short-read technology from Illumina and recently also the single-molecule real-time (SMRT) long-read sequencing technique from Pacific Biosciences. As a result, for some organisms numerous and diverse data sources are available. Combining these data into one high quality assembly still is a challenging task.
For the Chinese hamster, Cricetulus griseus, data is available from (i) Illumina short-read data from whole-genome libraries (including mate-pair libraries from varying insert sizes), (ii) Illumina short-read data from libraries of separated chromosomes, and (iii) long-read data generated with PacBio's sequencing technology. In order to get a genome assembly as good as possible, we compared and rated different assembly strategies taking into account technical conditions such as computing power needed and total computing times. For this comparison, we used four different de novo assemblies and further on combined these assemblies in different orders to form meta-assemblies.
Dipl.-Inf. Oliver Rupp, AG Bioinformatik und Systembiologie, Justus-Liebig-Universität Gießen
Wir stellen den neuen Stern an Himmel der Datenanalysesprachen vor, der angetreten ist, Matlab, R, Python & Co. in den Ruhestand zu schicken.
Ob er das Zeug dazu hat, kann jeder nach dem Vortrag für sich entscheiden.
Prof. Dr. Andreas Dominik, Fachbereich MNI, Technische Hochschule Mittelhessen, Gießen
Our current decade is strongly associated with the term Big Data, which attracts attention in many different fields, e.g., genomics, meteorology, or complex physical simulations. For instance, next-generation sequencing technologies are able to generate millions or billions of reads, which enabled completely new ways for precision medicine, in particular by using statistical and machine learning approaches. However, many new problems arise due to the huge amount of data generated. Computational methods needs to be adapted to address these big data issues and to path the way for precision medicine. As an example, recent developments in HIV diagnostics and therapy will be shown and discussed.
Prof. Dr. Dominik Heider, Heiderlab, Mathematik und Informatik, Philipps-Universität Marburg, Marburg
"SCOTCH: Subtype A Coreceptor Tropism Classification in HIV-1"
The V3 loop of the gp120 glycoprotein of the Human Immunodeficiency Virus 1 (HIV-1) is considered to be responsible for viral coreceptor tropism. gp120 interacts with the CD4 receptor of the host cell and subsequently V3 binds either CCR5 or CXCR4. Due to the fact that the CCR5 coreceptor is targeted by entry inhibitors, a reliable prediction of the coreceptor usage of HIV-1 is of great interest for antiretroviral therapy. Although several methods for the prediction of coreceptor tropism are available, almost all of them have been developed based on only subtype B sequences, and it has been shown in several studies that the prediction of non-B sequences, in particular subtype A sequences, are less reliable. Thus, the aim of the current study was to develop a reliable prediction model for subtype A viruses. Our new model SCOTCH is based on a stacking approach of classifier ensembles and shows a significantly better performance for subtype A sequences compared to other available models. In particular for low false positive rates (between 0.05 and 0.2, i.e., recommendation in the German and European Guidelines for tropism prediction), SCOTCH shows significantly better prediction performances in terms of partial area under the curves and diagnostic odds ratios compared to existing tools, and thus can be used to reliably predict coreceptor tropism for subtype A sequences.
"UROPA_GUI: a tool for Universal RObust Peak Annotation"
The annotation of genomic ranges such as peaks resulting from ChIP-seq/ATAC-seq or other experimental techniques represents a fundamental task for bioinformatics analysis with crucial impact on many downstream analyses. In our previous work, we introduced the Universal Robust Peak Annotator (UROPA), a flexible command line based tool which considerably extends the functionality of existing annotation software. In order to reduce the complexity for biologists and clinicians, we have implemented an intuitive web-based graphical user interface (GUI) for UROPA. This extension will empower all users to generate dynamic and specific annotation for a wide range of experimental setups considering reference feature type, position, orientation, and precedence.
Parastoo Fazelzadeh, PhD, Max-Planck-Institut für Herz- und Lungenforschung, Bad Nauheim
"De novo transcriptome assembly and training of support vector machine classifiers for prediction of antimicrobial peptides"
Mis- and overuse of antibiotics as well as the "new antibiotic paradoxon" have been leading to an increase in bacterial restistance against antibiotics and stagnation in new antibiotic development. An alternative to those classical antibiotics are antimicrobial peptides (AMPs). They are highly specific for prokaryotic cells and co-evolving in nature apart from synthetical derivates. With machine learning methods, it is possible to predict functional sequences. This was performed by support vector machines with preliminary principle component analysis. In parallel, a de novo transcriptome assembly was done for healthy and infected groups of Hirudo verbana (leech) with the goal to find new AMPs via comparative transcriptome analysis.
"Algorithmische Lösungen für den Umgang mit Big Data und Tiniest Devices"
Aufgrund von Big Data mit beispielsweise Anwendungen in der Bioinformatik sowie der Verwendung von kleinsten Geräten mit einem kleinen Speicher gibt es ein verstärktes Interesse an der Entwicklung platzeffizienter Algorithmen, d.h. Algorithmen mit einem Arbeitsplatzverbrauch, welcher kleiner ist als der der Standardalgorithmen für das betrachtete Problem. Der Vortrag gibt eine Übersicht der existierenden platzeffizienten Algorithmen für grundlegende Graphenprobleme und zeigt anhand des Beispiels von Tiefensuche wie Algorithmen platzeffizient gemacht werden können ohne dass dies zu wirklichen Laufzeiteinbußen führen muss.
Dr. habil. Frank Kammer, Fachbereich MNI, Technische Hochschule Mittelhessen, Gießen
"Bioinformatik als Data Science: Wie quetscht man das Wissen aus den Daten?"
Wer als Biologie, Ökologe oder Mediziner mehr über das Leben und die in ihm wirkenden Mechanismen herausfinden will, ist als empirischer Forscher natürlich auf Daten angewiesen. Mit neuen Methoden zur Datenerhebung ist es inzwischen möglich, schnell und kostengünstig große Mengen an Daten zu erheben. Beispielsweise produziert ein einzelner moderner DNA-Sequenzierer pro Woche das Hundertfache der Datenmenge, die man zur Jahrtausendwende zur Entschlüsselung des menschlichen Genoms gesammelt hatte, damals noch mit großem Aufwand und nach jahrelanger Arbeit. Während es also in weiten Bereichen der Biowissenschaften immer leichter wird, Daten in ausreichender Menge zu sammeln, wird es gleichzeitig immer schwerer, diese Daten angemessen auszuwerten. Die seit Jahrzehnten exponentiell anwachsende Datenflut stellt die Bioinformatik vor eine große Herausforderung. In diesem Vortrag werde ich anhand von Beispielen aus meinen Forschungsprojekten der letzten Jahre zeigen, wie die angewandte Bioinformatik durch Kombination unterschiedlicher Daten versucht, neue Einblicke in biologische Prozesse zu gewinnen.
Dieser KoBIS-Vortrag ist gleichzeitig Teil der Ringvorlesung des Masterstudiengangs „Bioinformatik und Systembiologie“. Studierende der Bioinformatikstudiengänge erhalten einen Einblick in mögliche Themen für Laborpraktika oder Abschlussarbeiten.
Prof. Dr. Andreas Gogol-Döring, Fachbereich MNI, Technische Hochschule Mittelhessen, Gießen
"TAPscan: Accurate genome-wide gene family annotation and web representation"
Transcription associated proteins (TAPs) are major players in gene regulatory networks and contribute to increasing the potential complexity of gene network circuitry. We have updated the workflow established by Lang et al. (2010) consisting of a set of domain-based classification rules aimed to identify plant transcription factors (TFs), transcriptional regulators (TRs) and putative (in silico predicted) TAPs (PTs) amongst a given set of proteins. With a combination of custom built and existing Hidden Markov Model (HMM) domain profiles a total of 122 TAP families can now be distinguished. We have also developed a web interface that can be used to browse through and download our results. The setup of the web interface has been designed with the aims to maximize applicability and alleviate maintenance.
Cornelia Mühlich, M. Sc. u. Per Wilhemsson, M.Sc., Rensing lab, Zellbiologie, Philipps-Universität Marburg, Marburg
"Stable Isotope Tracing Models to understand elemental cycling in Terrestrial ecosystems"
Gross elemental transformation rates in terrestrial ecosystems are typically quantified via stable isotope tracing techniques. Basics of stable isotope chemistry and the development of suitable numerical stable isotope models are a prerequisite for successful quantification. In this presentation development of a suitable numerical tracing techniques will be presented and applications in MatLab and Simulink will be discussed.
Prof. PhD Christoph Müller, Institut für Pflanzenökologie, Justus-Liebig-Universität Gießen
"Classification of Normal/Abnormal Heart Sound Recording: the PhysioNet/Computing in Cardiology Challenge 2016"
The 2016 PhysioNet/CinC Challenge aims to encourage the development of algorithms to classify heart sound recordings collected from a variety of clinical or nonclinical (such as in-home visits) environments. The aim is to identify, from a single short recording (10-60s) from a single precordial location, whether the subject of the recording should be referred on for an expert diagnosis.
Prof. Dr. Andreas Dominik, Fachbereich MNI, Technische Hochschule Mittelhessen, Gießen
"Recognition of Abnormalities in Heart Sound Recordings by Reconstruction of Idealised Beats"
We present algorithms to distinguish between healthy and diseased condition of the heart, based on the analysis of phonocardiograms. The software tries to mimic the decision-making process of a cardiologist by identifying heart beats (S1 and S2), finding extra sounds and murmurs while ignoring all kinds of artefacts and noise.
Two different solutions have been submitted to the PhysioNet Challenge 2016: The entry for phase I aims to reconstruct the signal of an ideal heartbeat by calculating the median of an overlay of all beats of a recording. An LVQ-classifier, trained with the ideal beat of 3240 PCGs of the challenge training set, achieved a specificity of 0.85 and a sensitivity of 0.40, resulting in a total score of 0.63.
Our entry for the official phase of the challenge searches for abnormalities in every single beat of a PCG. The results display a sensitivity of 0.91, a specificity of 0.29, and a total score of 0.60.
Simon Hofmann, M.Sc., Fachbereich GES, Technische Hochschule Mittelhessen, Gießen
"Using Deep Gated RNN with a Convolutional Front End for End-to-End Classification of Heart Sound"
Classification of heart sounds of a diverse set of phonocardiograms (PCGs) from different recording settings is the challenging objective of the 2016 PhysioNet Challenge. We suggest an end-to-end deep neural network, which is fed with raw PCGs and which learns to autonomously extract features and to classify the recordings. Our architecture combines convolutional and recurrent layers, followed by an attention mechanism, which weights time steps by importance and a dense multilayer perceptron as classifier.
Whereas currently trending deep neural networks in speech recognition or computer vision use up to a million of training samples, a restricted set of only 3,153 heart sound recordings is available as training data. We workaround this limitation by artificially increasing the training set by means of augmentation of the raw PCGs using various audio effects.
Using this moderately sized neural network, we attain high validation scores of 0.89 on validation data; however the resulting scores on the hidden test data of the challenge diverge in range (0.82).
Christian Thomae, B.Sc., Fachbereich MNI, Technische Hochschule Mittelhessen, Gießen
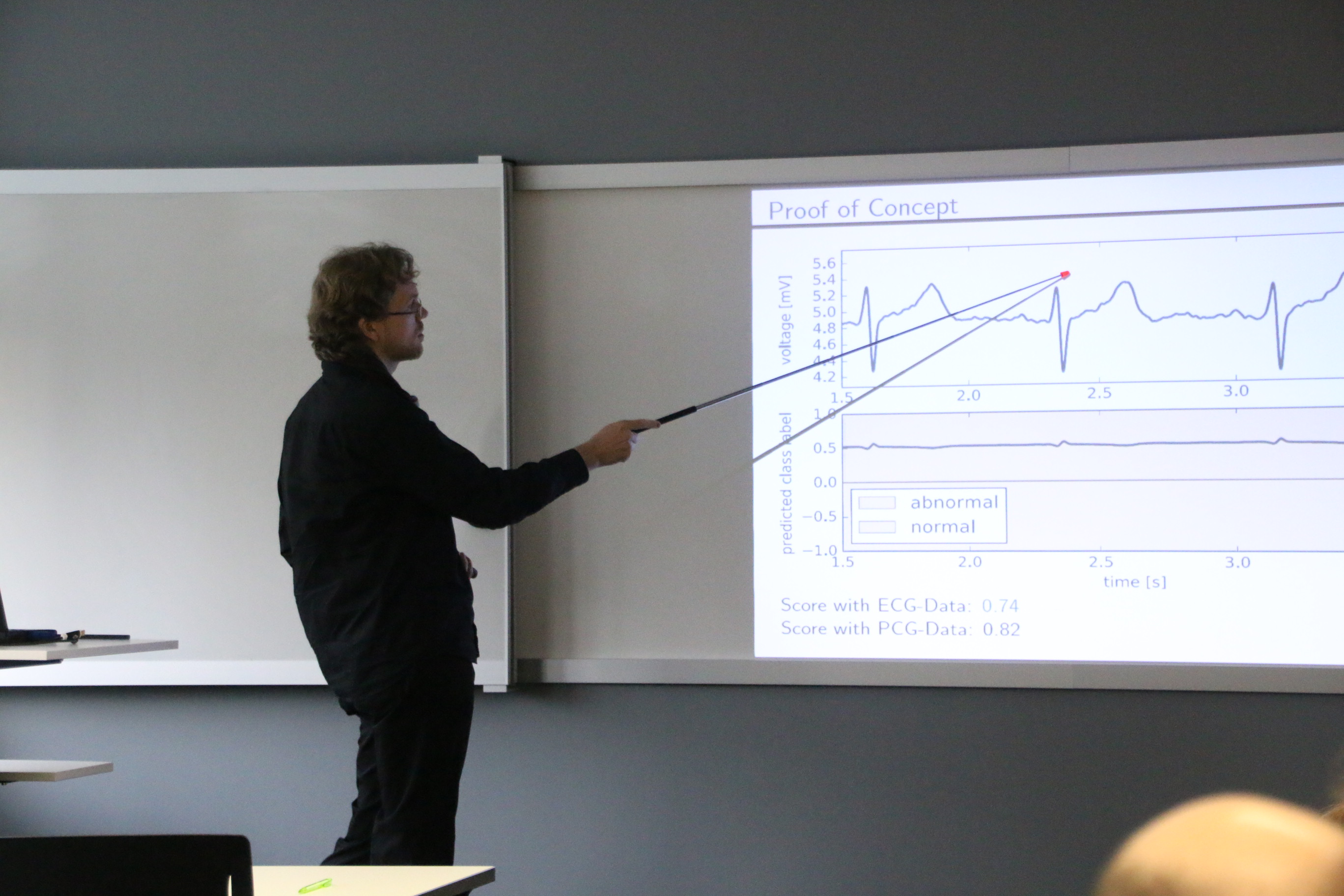
"Can Electrocardiogram Classification be Applied to Phonocardiogram Data? – An Analysis Using Recurrent Neural Networks"
Both a Phonocardiogram (PCG) and an Electrocardiogram (ECG) are sequential measurements of heart activity used to distinguish normal from abnormal heart function. Although they measure different physical quantities, we show that training a long short-term memory network on the Physionet challenge using only the ECG data available for the MIT heart sounds database still yields a score of 0.74 compared to the reference score of 0.82 for a similar net trained on the PCG data.
This finding suggests that it may be valuable to train a transformational neural network to produce an artificial ECG from a PCG. Such a transformational net would allow to harness the know-how of decades of research on ECG classification to improve PCG classification. Unfortunately, this task seems too hard for current state-of-the art architectures for neural networks given the data of the Physionet challenge 2016. However, it may be worthwhile to further pursue this approach using data with less variance in the ECG signals or a specialized network architecture.
Christopher Schölzel, M.Sc., Fachbereich MNI, Technische Hochschule Mittelhessen, Gießen
"Using different combinations of Illumina short read data and PacBio long read data for the de novo assembly of the Chinese hamster genome"
Technical developments in next-generation sequencing as well as decreasing prices have enabled various sequencing projects including the large genomes of higher eukaryotes. The two most popular and efficient methods for whole-genome sequencing are the short-read technology from Illumina and recently also the single-molecule real-time (SMRT) long-read sequencing technique from Pacific Biosciences. As a result, for some organisms numerous and diverse data sources are available. Combining these data into one high quality assembly still is a challenging task.
For the Chinese hamster, Cricetulus griseus, data is available from (i) Illumina short-read data from whole-genome libraries (including mate-pair libraries from varying insert sizes), (ii) Illumina short-read data from libraries of separated chromosomes, and (iii) long-read data generated with PacBio's sequencing technology. In order to get a genome assembly as good as possible, we compared and rated different assembly strategies taking into account technical conditions such as computing power needed and total computing times. For this comparison, we used four different de novo assemblies and further on combined these assemblies in different orders to form meta-assemblies. The evaluation of the four assemblies and the four meta-assemblies relied on 65 different metrics that include parameters such as the total genome size, the number of contigs and scaffolds, and the correct allocation of the contigs to the single chromosomes. It showed that the meta-assemblies improved the initial assemblies in terms of contig and scaffold length, the N50, the L50 numbers, the gene content, and the sequence quality. Furthermore our results suggest that a meta-assembly that is started from an assembly computed with PacBio data outperforms meta-assemblies that are computed on the basis of Illumina assemblies.
Dipl.-Inf. Oliver Rupp, AG Bioinformatik und Systembiologie, Justus-Liebig-Universität Gießen
"MGX - A flexible metagenome analysis framework"
The characterization of microbial communities based on sequencing and analysis of their genetic information has become a popular approach also referred to as metagenomics; in particular, the recent advances in sequencing technologies have enabled researchers to study even the most complex communities consisting of thousands of species. Metagenome analysis, the assignment of sequences to taxonomic and functional entities, however, remains a tedious task: large amounts of data need to be processed. There are a number of approaches that aim to solve this problem addressing particular aspects, however, scientific questions are often too specific to be answered by a general-purpose method. For this reason, we developed MGX as an extensible framework for the management and analysis of metagenomic datasets; MGX provides a complete set of workflows required for taxonomic and functional metagenome analysis. In contrast to existing platforms, MGX is easily extendable and allows researchers not only to execute predefined but also custom analysis pipelines within its framework. MGX enables fast adoption of novel algorithms e.g. for the taxonomic classification of metagenomic sequences, thereby offering researchers to use the most current tools for the analysis of their datasets.
Dipl.-Inf. Sebastian Jaenicke, AG Bioinformatik und Systembiologie, Justus-Liebig-Universität Gießen
"Stem cell models in preclinical and clinical setting: current status and future prospects in healthcare system"
Dr. rer. nat Shibashish Giri, Applied Stem Cell Biology and Cell Technology, Biomedical and Biotechnological Center (BBZ), Medical faculty, University of Leipzig
"Metabolic lifestyle and energy conservation tricks of giant, symbiotic bacteria ― the special case of Epulopiscium"
Research in the last few decades has brought considerable advances in our understanding of the biology of several giant bacteria that seem to bypass the diffusional limitations of unicellular prokaryotes lacking specialized intracellular transport mechanisms, including two marine sulfur-oxidizing species (Beggiatoa and Thiomargarita) and the gut symbiont of herbivorous surgeonfishes, Epulopiscium fishelsonii. The latter bacteria are morphologically diverse, sometimes longer than 600 µm, and extraordinarily polyploidic, carrying as much as 600,000 genome copies per cell. Unlike other bacteria, they form multiple intracellular offspring in a circadian reproductive cycle that is synchronized with the feeding behaviour of their hosts. However, very little is known about the metabolic traits of Epulopiscium and their potential roles in the digestive process. In this talk, I will provide genomic evidence that links metabolism, lifestyle, and mechanisms for energy-conservation in Epulopiscium and advances our understanding of their potential physiological roles in their symbiosis with surgeonfishes.
Dr. David Kamanda Ngugi, Red Sea Research Center, King Abdullah University of Science and Technology (KAUST), Thuwal, Kingdom of Saudi Arabia
"Introduction to the IT infrastructure of the Bioinformatics Core Facility (BCF)"
The Bioinformatics Core Facility (BCF) hosts a centralized IT-infrastructure for all bio-computational services, storage, computing and specialized hardware systems. The facility also provides a comprehensive bioinformatics-IT-environment, software support and automated pipelines for batch processing of typical high-throughput workflows, a collection of bioinformatics databases and other ressources. Furthermore we offer bioinformatics teaching and training, consulting and support for our partners and research collaborations.
Dr. Burkhard Linke u. Dr. Marc Bruckskotten, AG Bioinformatik und Systembiologie, Justus-Liebig-Universität Gießen
"High-throughput software pipelines for genotypic antimicrobial resistance testing"
Das Ziel der Arbeit war die Erstellung eines Programms zur Erkennung verschiedener Antibiotikaresistenzen in S. aureus. Ausgehend von publizierten resistenzverursachenden Mutationen wurde die Implementierung mit 924 Testfiles überprüft und optimiert und eine Erkennungsrate innerhalb der FDA Limits erzielt.
Jonas Sträßer, Fachbereich MNI, Technische Hochschule Mittelhessen, Gießen
"Strategies to identify genomic signatures of allopolyploidisation in Brassica napus"
Allopolyploidization leads to the formation of new species with the potential of fast adaptation to rapidly changing environmental conditions and plays therefore an important role in plant diversification. The genome of the allopolyploid plant species Brassica napus (AACC, 2n=38; oilseed rape, canola) derived from the interspecific hybridization of the ancestral hexaploid genomes of B. rapa (AA, 2n=20) and B. oleracea (CC, 2n=18). Synthetic combination of the two diploid genomes leads to chromosomal rearrangements induced by meiotic errors during the first generations after hybridization, potentially creating variation which is relevant for both natural and artificial selection. In my talk I will describe a data analysis pipeline to identify chromosome restructuring patterns in polyploid genomes based on whole-genome resequencing datasets. Using this method we investigated polyploidisation signatures in 51 diverse natural and synthetic accessions of Brassica napus.
Dr. Birgit Samans, Institut für Pflanzenbau und Pflanzenzüchtung, Justus-Liebig-Universität
"Differentielle Genexpressionsanalyse von in vivo markierter RNA / small-RNA-Seq Daten der Honigbiene: Eine Zeitreihenanalyse"
Die Honigbiene (Apis mellifera) ist ein Schlüsselmodell für hoch soziale
Spezies. Ihre sehr komplexen Verhaltensstrukturen, einschließlich Lernen und Gedächtnisbildung, sowie die Reaktion auf verschiedene Stressfaktoren, können auf molekularer Ebene als Änderung in der Genexpression gesehen werden. Die Genexpression wird durch die Aktivierung von Transkriptionsmaschinerie und Chromatinstruktur reguliert. Auf post-transkriptionaler Ebene erfolgt die Regulation durch kurze, hoch konservierte, nicht codierende RNAs (miRNA).
Um verschiedene Fragestellungen ansprechen zu können, adaptierten und wendeten wir eine Technik an, die eine in vivo Markierung von de novo transkribierter mRNA in einem definierten Zeitfenster erlaubt. So erfolgten bereits eine Reihe differentieller Genexpressionsstudien auf Basis von Lernexperimenten.
Schwerpunkt meiner Arbeit ist die Etablierung einer Pipeline zur Analyse von Zeitreihen RNA- / small-RNA Sequenzierdaten.
Marie-Theres Arend, M.Sc., AG Zoologie/Physiologie-Neurobiologie , Universität des Saarlandes
"Efficient analysis of NGS data by workflow based systems"
Current chromatin research often involves the application of next generation sequencing (NGS)-based methods in order to gain structural as well as functional insights into how the chromatin template exerts its regulatory effects on nuclear processes like transcription, replication or DNA repair. Analysis of NGS data comes along with the requirements for computational infrastructure that allows the execution of relevant analysis tools. In order to enable our co-workers to perform analysis of their NGS-based data sets without expert programming knowledge we would like to provide an integrated workflow environment system. The workflow environment will be accessible to experimental biologists in order to enable them to perform standard ChIP-seq, RNA-seq and other chromatin-related data analysis workflows that are based on NGS-related techniques (MNase-seq, "3C"-based methods, etc.). In addition to basic workflows for execution of principal analysis steps like mapping, peak calling and annotation as well as visualization for exploratory analysis we would like to integrate more specific and complex workflows dedicated to project-specific questions. For this purpose we have decided to set up a GALAXY server, which allows maximum flexibility with respect to integration of already existing tools as well as extensibility in order to include additional more and specific data analysis tools. In example the Conveyer workflow engine will be used to sum up complex analysis steps to reduce the complexity and time of NGS analysis.
Tobias Zimmermann, M.Sc., AG Bioinformatik und Systembiologie, Justus-Liebig-Universität
"Analysis of newborn screening data: New approaches in detecting metabolic disorders"
Newborn screening is applied to every newborn baby within the first 48 hours of life and includes measurement of 92 metabolies and aminoacids. The aim of this screening is to detect diseases as early as possible. Screening results do not confirm a disease, but identify abnormal values. More specific tests are then used to determine if a disease condition is present.
There are 12 disease conditions which are covered by the current screening. Most of them are rare metabolic diseases. About 1 in 1623 children suffers from one of these. But in the screening about every second child shows at least one abnormal value. To improve this high false-positive rate a dataset of 20,000 anonymised screenings results were evaluated with different approaches. The primary focus was to build a basis for the selection of cut-off values, which determine what is labeled as abnormal. Furthermore, it was evaluated whether a multi-dimensional analysis, with the usage of machine-learning, can support the identification of special conditions.
In this presentation methods and results of this analysis will be displayed.
Michael Menzel, B.Sc., Fachbereich MNI, Technische Hochschule Mittelhessen, Gießen
Beim "Bio Entity Graph Visualizer" handelt es sich um eine Visualisierung von Relationen zwischen Datenbankeinträgen verschiedener biologischer Strukturen anhand von gerichteten Graphen. Der Fokus liegt auf einer effizienten Strukturierung und Darstellung der vorhandenen Daten, Usability sowie Erweiterbarkeit.
Ellen Marhold, Fachbereich MNI, Technische Hochschule Mittelhessen
"Genome Analyzer - Präsentation des Softwaretechnik-Praktikums im Wintersemester 2015/16"
Die Analyse von Integrationsstellen – Positionen im Genom, an denen fremde DNA eingefügt wird – ist ein wichtiges Forschungsgebiet, um beispielsweise Viren typisieren zu können. Mit den üblichen Genomebrowsern lässt sich die Analyse sehr vieler dieser Positionen innerhalb eines Genoms jedoch nicht bewerkstelligen. Daher entwickelten Bioinformatik-Studierende innerhalb des Softwaretechnik-Praktikums den "Genome Analyzer", welcher nicht einzelne Positionen anzeigt, sondern Statistiken über die Charakteristik der Integrationsstellen erstellt. Das achtköpfige Team nutzte dazu Technologien und Tools wie Java, Maven, Git und Redmine. Ihr persönliches Fazit und das fertige Produkt werden die Studierenden beim Abschluss des Projekts am 01. März vorstellen.
Studenten des Softwaretechnik-Praktikums, Fachbereich MNI, Technische Hochschule Mittelhessen
"Data preparation for transcriptome analysis with microarrays"
Transcriptome analysis has become more and more important in the development of diagnostics and the search for biomarkers. Microarray analysis bears the advantage to analyze a great number of transcripts simultaneously and facilitate a better understanding of the role and involvement of specific transcripts. It may provide hints to biological mechanisms resulting from the gene expression profile or underlying mechanisms resulting in the observed gene expression profiles. To detect meaningful gene expression it is important to assure data quality in the preprocessing of microarray data.
A workflow for data preprocessing and preparation has been developed for the open-source platform in R (r-project.com) with the help of R- and Bioconductor packages. It will be shown, that preprocessing plays an important role in the discovery and correction of handling errors, e.g., mislabeled arrays.
Dr. Anita Windhorst, Institut für Medizinische Informatik, Justus-Liebig-Universität Gießen
"A microarray study based on tracheal aspirate fibroblasts with regard to treatment of severe lung diseases in premature born infants"
Microarray analysis is a powerful tool for simultaneously screening a high amount of transcripts resulting for example from different disease states or treatment variants. Statistical analysis of different experiments or conditions results in sets of significantly differentially regulated genes. In order to develop a hypothesis about gene function or functional changes, an in depth analysis of the resulting gene sets has to be conducted by applying diverse strategies and tools. This procedure will exemplary be shown by presenting a micro array study based on tracheal aspirate fibroblasts with regard to treatment of severe lung diseases in premature born infants.
Application of glucocorticoids in preterm infants is aimed at reducing risk of inflammation, since in these patients high oxygen concentrations immediately after delivery are associated with elevated cytokine plasma levels and bronchopulmonary dysplasia (BPD). The glucocorticoid receptor (GR), which predominantly mediates the effect of glucocorticoid (GC) is involved in many homeostasis regulating processes and diverse stress pathways. The application of artificial glucocorticoid – dexamethasone (DEX) – on cultivated tracheal aspirate fibroblasts has been used to infer genes, which are involved in these processes. The results of this study will be presented.
Dr. Melanie Markmann, Medizinische Mikrobiologie, Justus-Liebig-Universität Gießen
"Modeling Biology in Modelica: The Human Baroreflex"
Modelica is a declarative object-oriented acausal programming language designed for modeling complex physical systems. Using the example of a model of the human baroreflex, we show that this language can produce the same simulation results as the reference implementation in C. With our implementation we demonstrate that Modelica is perfectly suited to for building biological systems in a natural representation.
Christopher Schölzel, M.Sc., Fachbereich MNI, Technische Hochschule Mittelhessen, Gießen
"Silicon Heart: An Easy to Use Interactive Real-Time Baroreflex Simulator"
A simulator of the baroreflex loop is implemented as a distributed system, including independent functional units with each of them running without synchronisation and in real-time. Individual components are build from extended equations of the well established Seidel-Herzel-model.
The system includes five small computers representing five independent sub-models. Each component has a computer mouse connected that allows for real-time manipulation of simulation parameters in the respective part of the model. This way, numerical values of variables, such as neurotransmitter concentrations or breathing frequency, can easily be altered by turning the associated adjusting wheel.
Virtual administration of drug substances and virtual disease simulations are performed and show that the asynchronous simulation is robust enough to be used as an intuitive model to study heart rate dynamics.
Michael Menzel, B.Sc., Fachbereich MNI, Technische Hochschule Mittelhessen, Gießen
"Modeling the kinetics of the cardiac acetylcholine receptor M2"
The muscarinic acetylcholine recepor M2 located at the heart muscle cells is modeled using Modelica. The model includes the receptor, the different stages of the G-protein and the decomposition of acetylcholine by acetylcholinesterase.
"Bryophyte-specific orphan genes inferred from the onekp project"
Here, we analyzed transcript data from 7 hornworts, 26 liverworts and 41 mosses from the 1KP project (www.onekp.com) to determine the origin of genes and the abundance of genome duplication events in bryophytes. Sequence comparisons of the transcriptome data with published plant genomes and a representative set of genomes outside the green tree of life were not only used to construct phylostratigraphic gene age maps, but also to infer orthologous groups within and between the analyzed bryophyte species.
Dr. Kristian Ullrich, Rensing lab, Zellbiologie, Philipps-Universität Marburg, Marburg
"Gene family classification using HMM domain-based rule sets"
Using profile Hidden Markov Models (HMMs) proteins can be scanned for domains. Combining the result with domain-based rule sets makes it possible to classify proteins into different gene families based on their domain architecture.
Per Wilhemsson, M.Sc., Rensing lab, Zellbiologie, Philipps-Universität Marburg, Marburg
"Reproducibility and normalization issues of RNA-seq experiments"
The number of RNA-seq data in the databases rises quickly. To analyze and compare the different data sets, normalization is indispensable. Not only digital normalization is the method of choose. Adding spike-ins to your library could also be an interesting option.
"Computergestützte Annotation von Aminosäuresequenzen am Beispiel von rekombinanten Antikörpern und CARs (Chimeric Antigen Receptors)"
In der Arzneistoffentwicklung werden sämtliche Sequenzen registriert und in Datenbanken gelagert, um auch anderen Forschern zur Verfügung zu stehen. Die Pflege solcher Datenmengen ist eine mühevolle Aufgabe. Daher kommen für die Annotationen dieser Sequenzen computergestützte Services zum Einsatz. An Beispielen der aktuellen Antikörperforschung wird der Aufbau, die Funktionsweise und die Erweiterung eines solchen Sequence Annotation Service vorgestellt.
"Genzentrierte Datenvisualisierung von TCGA Daten"
Seit Beginn des Projekts „The Cancer Genome Atlas“ wurden Genom-Daten von über 30 Krebsgruppen erhoben und der Öffentlichkeit zur Verfügung gestellt. Für die weitere Analyse der Datensätze wurde ein Genzentrischer Target CV aus den unter anderem eruierten mRNA Expressions-Werten, Kopierzahlvariationen und DNA-Änderungen generiert.
"Global Data Share - Improving research in the field of oncology by providing easy access to omic data. The first part of the project phase, completed tasks and prospects for the upcoming Bachelor Thesis."
Sebastian Spänig, Fachbereich MNI, Technische Hochschule Mittelhessen, Gießen
"EDGAR 2.0: an enhanced software platform for comparative gene content analyses"
The deployment of next generation sequencing approaches has caused a rapid increase in the number of completely sequenced genomes. As one result of this development, it is feasible to analyze not only single genomes, but large groups of related genomes in a comparative approach. A main task in comparative genomics is the identification of differential gene content in a number of genomes, i.e., the core genome or singleton genes. To support these studies the EDGAR software was developed. Using a generic orthology criterion based on the distribution of alignment hits within a genus EDGAR is designed to automatically perform genome comparisons in a high throughput approach. Comparative analyses for 2072 genomes across 161 genera taken from the NCBI genomes database were conducted with the software and the results are provided as a free-to-use public database. EDGAR provides several analysis and visualization features and significantly simplifies the comparative analysis of related genomes. The web-based user interface offers Venn diagrams, synteny plots, and a comparative view of the genomic neighbourhood of orthologous genes. Recently, the software was extended with various new features like statistical and phylogenetic analyses, replicon grouping options and second level analyses of meta-genesets. Noteworthy, EDGAR calculates phylogenetic trees as well as amino acid identity (AAI) matrices based on all genes of the core genome, providing a solid basis for both analyses. Thus the software supports a quick survey of evolutionary relationships and simplifies the process of obtaining new biological insights into the differential gene content of kindred genomes. EDGAR is among the most established tools in the field of comparative genomics. In addition to the publicly available projects more than 150 private projects with more than 5000 analyzed genomes have been computed during the last 5 years. EDGAR is available via the public web server http://edgar.computational.bio.
Dr. Jochen Blom, AG Bioinformatik und Systembiologie, Justus-Liebig-Universität Gießen
"Viral genetic diversity from next-generation-sequencing-data: Challenges and Opportunities from the bioinformatics point of view"
RNA viruses, exist in large populations and display high genetic heterogeneity within and between infected hosts. Assessing intra-patient viral genetic diversity is essential for understanding the evolutionary dynamics of viruses, for designing effective vaccines, and for the success of antiviral therapy. Next-generation sequencing (NGS) technologies allow the rapid and cost-effective acquisition of thousands to millions of short DNA sequences from a single sample. However, this approach entails several challenges in terms of experimental design and computational data analysis. Although the massively parallel sequencing approach can detect low-frequency mutations and it provides a snapshot of the entire virus population, analyzing deep sequencing data obtained from diverse virus populations is challenging because of PCR and sequencing errors and short read lengths, such that the experiment provides only indirect evidence of the underlying viral population structure. Recent computational and statistical advances allow for accommodating some of the confounding factors, including methods for read error correction, haplotype reconstruction, and haplotype frequency estimation. In this talk we will give an overview of the state-of-the-art in this field and review opportunities and shortcomings of the statistical models and algorithms involved and discuss their implications of these shortcomings in terms of experimental design and sequencing strategies.
Prof. Dr. Franz Cemic, Fachbereich MNI, Technische Hochschule Mittelhessen, Giessen
"de.NBI - Das Deutsche Netzwerk für Bioinformatik-Infrastrukturen"
Das BMBF finanziert das „Deutsche Netzwerk für Bioinformatik-Infrastruktur“ (de.NBI) bis 2020 mit insgesamt 22 Millionen Euro. In dem Netzwerk arbeiten acht deutsche Zentren zusammen, die für die Bearbeitung bioinformatischer Daten auf dem Gebiet der Lebenswissenschaften ausgewiesen sind. Als Gemeinschaftseinrichtung bieten sie künftig bioinformatische Dienstleistungen für Forschungsprojekte aus Biotechnologie und Biomedizin an. Außerdem bildet das Netzwerk Forscherinnen und Forscher in der Nutzung von Bioinformatik-Software aus. In meinem Vortrag gebe ich einen Überblick über das Deutsche Netzwerk für Bioinformatik-Infrastruktur und beschreibe die Aufgaben und Zielsetzungen des Leistungszentrums BiGi - Bielefeld-Gießen Zentrum für Mikrobielle Bioinformatik.
Prof. Dr. Alexander Goesmann, AG Bioinformatik und Systembiologie, Justus-Liebig-Universität Gießen
"Glycosciences.de und MonosaccharideDB: Glyco-Bioinformatik in Gießen"
Die Glyco-Bioinformatik ist ein recht junges Teilgebiet der Bioinformatik. Im Fokus stehen hier Kohlenhydrate sowie Glycokonjugate (insb. Glycoproteine und Glycolipide) sowie die Wechselwirkungen zwischen Kohlenhydraten und anderen Molekülen. Diese Wechselwirkungen spielen eine wichtige Rolle bei der Kommunikation zwischen Zellen, aber auch bei verschiedenen Krankheiten sowie der Immunantwort. Das Gießener Glycosciences.de Portal ist eines der größten Glyco-Bioinformatik-Portale weltweit. Der Hauptfokus liegt hier auf der Modellierung und Analyse von 3D-Strukturdaten, wobei die Protein Data Bank (PDB) eine wichtige Datenquelle ist.
Ein Hindernis bei der weltweiten Entwicklung der Glyco-Bioinformatik ist die geringe Vernetzung der verschiedenen Datenbanken und Programme. Der Austausch von Daten sowie die Querverlinkung der Datenbanken scheitern häufig schon an unterschiedlichen Sequenzformaten zur Speicherung der Kohlenhydrate. Bei der Konvertierung dieser Formate müssen insbesondere auch die Namen für die einzelnen Bausteine, die Monosaccharide, übersetzt werden. Hierzu wurde die MonosaccharideDB entwickelt, die ebenfalls in Gießen gepflegt wird. Sie fungiert als Referenzdatenbank zur Monosaccharid-Nomenklatur und ist dabei so aufgebaut, dass nicht nur die zurzeit knapp 800 vorgespeicherten Monosaccharide abgefragt werden können, sondern über Parser- und Encoder-Routinen auch zu noch nicht gespeicherten Bausteinen Informationen „on the fly“ bereitgestellt werden können. Damit liefert MonosaccharideDB einen wichtigen Beitrag bei der weltweiten Vernetzung von Glyco-Bioinformatik-Ressourcen und somit zur weiteren Entwicklung der Glyco-Bioinformatik.
Dr. Thomas Lütteke, Institut für Veterinärphysiologie und Biochemie, Justus-Liebig-Universität Gießen
"BioLib - Präsentation des Softwaretechnik-Praktikums im Wintersemester 2014/15"
Ein essenzieller Bestandteil der Arbeit eines Bioinformatikers ist das Entwickeln von Software. Um für biologische Fragestellungen Software zu programmieren, ist das fundierte Hintergrundwissen der Lebenswissenschaft unersetzlich. Das musste die letzte Gruppe des Softwaretechnik-Praktikums erfahren, die mit nur einem Bioinformatiker in ihren Reihen eine Bibliothek für Sequenzalgorithmen weiterentwickelte. Unter Verwendung von Technologien und Tools wie Java, Git und Redmine führten sie ein umfassendes Refactoring der bereits bestehenden Bibliothek durch und erweiterten sie um einige Algorithmen wie etwa BLAST. Wie die 14-köpfige Gruppe ihre Arbeit organisierte, welche Hindernisse und Lösungen es gab und wie das endgültige Produkt aussieht, präsentieren die Studenten am 07.04.2015.
Studenten des Softwaretechnik-Praktikums, Fachbereich MNI, Technische Hochschule Mittelhessen
"Cardiovascular Bioinformatics: Perspective on long noncoding RNAs"
Dr. Shizuka Uchida, Institute of Cardiovascular Regeneration, Goethe-Universität Frankfurt
"C-It-Loci: A knowledge database for tissue-enriched loci"
Tyler Weirick, M.Sc., Institute of Cardiovascular Regeneration, Goethe-Universität Frankfurt
"RNAeditor: a tool to identify editing events from RNA-seq data"
Dipl.-Bioinf. David John, Institute of Cardiovascular Regeneration, Goethe-Universität Frankfurt
About:
Das Kolloquium für Bioinformatik und Systembiologie Mittelhessen (KoBIS) ist eine gemeinsame Initiative der Technischen Hochschule Mittelhessen und der Justus-Liebig-Universität Gießen zum wissenschaftlichen Austausch zwischen Arbeitsgruppen und Hochschulen, die sich mit dem Thema Bioinformatik und Systembiologie beschäftigen. Seit Oktober 2015 gehört auch die Phillips-Universität Marburg zu den regelmäßig teilnehmenden Hochschulen. Präsentiert werden Projekt- und Abschlussarbeiten sowie aktuelle Forschungsthemen von Mitarbeitern und externen Gästen.
Interessierte sind herzlich willkommen!
Logo:
Bei dem KoBIS-Logo handelt es sich um eine Abwandlung eines Modells von H. Vogel zur Beschreibung des Blüten- bzw. Samenstands der Sonnenblume. Es symbolisiert damit also die Verbindung zwischen Biologie und Informatik bzw. Mathematik. Für Interessierte gibt es auch noch eine längere Nerd-Variante dieser Erklärung.
Zum Erstellen von Präsentationsfolien oder Artikeln zu KoBIS gibt es das Logo natürlich auch als Vektorgrafik.